The following technology platforms are employed for our assay services or products:
Luminex Bead-Based Multiplex Assay
Our multiplexing immunoassays with bead suspensions are
based on xMAP (Multiple- Analyte
Profile) technology developed by Luminex Corp.
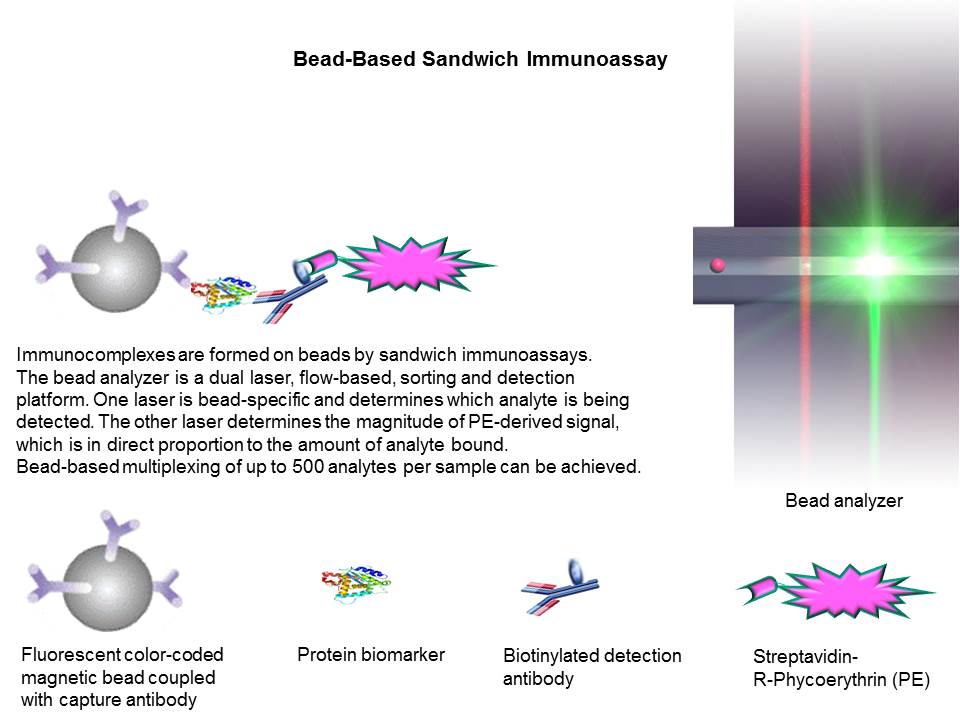
Luminex bead-based multiplexing sandwich
immunoassays for protein biomarkers employ a suspension of microsphere sets in
a 96-well microplate where each bead set represents an individual immunoassay.
The microspheres are 6.5-µm paramagnetic beads functionalized with carboxyl groups on the bead
surface for covalent attachment of capture antibodies. The beads are internally
encoded by fluorescent dyes and individually addressed to a specific assay in
the multiplexed tests. This technology uses a proprietary dying process to
create up to 500 unique dye mixtures which are used to identify an individual
bead. The fluorophore-coded beads function as bar codes for individual analyte. Each bead set can be coated with a capture
antibody specific to one analyte. During a bead-based
sandwich immunoassay, captured analyte on the bead
surface is bound by a biotinylated detection antibody and streptavidin-phycoerythrin (S-PE). Multiple analytes in a single
aliquot of serum, plasma, cell culture supernatant, cell lysate and tissue
extract, etc. are determined quantitatively and simultaneously with a Luminex bead reader. Within the bead reader, one laser
excites the beads’ internal dyes, which identify each bead set by its fluorescent
color code, while a second laser excites the reporter dye (PE) captured during
the assay. Tens to hundreds of readings are made for each bead set. The bead reader
classifies each bead according to its predefined map region by means of digital
signal processing. The magnitude of the reporter-derived signal is in
direct proportion to the amount of analyte bound. Analyte concentrations
of the multiplex immnuassays are typically determined
by 5-parametere logistic regression algorithm with analysis of the median
fluorescence intensity readings of each 8-point calibrator curve.
High-Sensitivity MSD-ECL Multiplex Assay
In addition to
conventional Luminex bead-based multiplex assays and ELISAs as a broad initial
analysis for biomarker profiling, our high-sensitivity singleplex
and multiplex assays are established upon Meso Scale Diagnostics-
electrochemiluminescence (MSD-ECL) technology platform to detect low-abundance
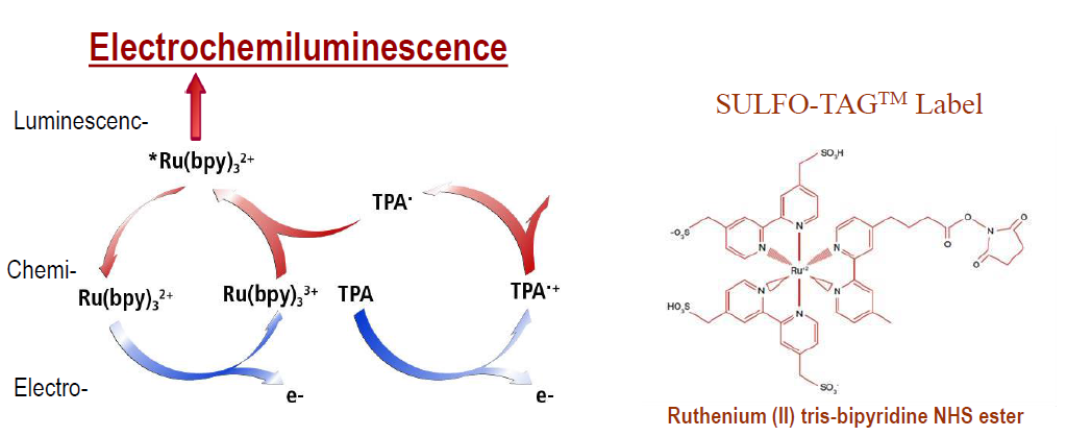
biomarkers difficult to measure with conventional assays. Electrochemiluminescent
labels generate light when stimulated by electricity in the appropriate
chemical environment. This reaction is incorporated into our immunoassays to
provide the light signal used to measure important proteins and other
biomedical molecules. Multiple excitation cycles can amplify signals
to enhance light levels. The stimulation method (electricity) is decoupled from
the signal (light) allowing only labels near the electrode surface to be
detected. MSD-ECL assay sensitivity increases over conventional assays result from
ECL signal amplification and low background (high ratio of signal-to-noise). Additionally,
the wider dynamic range (3-4+logs) of MSD-ECL detection systems can be achieved so that high and low
expression levels can be measured without multiple sample dilutions.
How does
electrochemiluminescence work?
Electrochemiluminescence Implementation
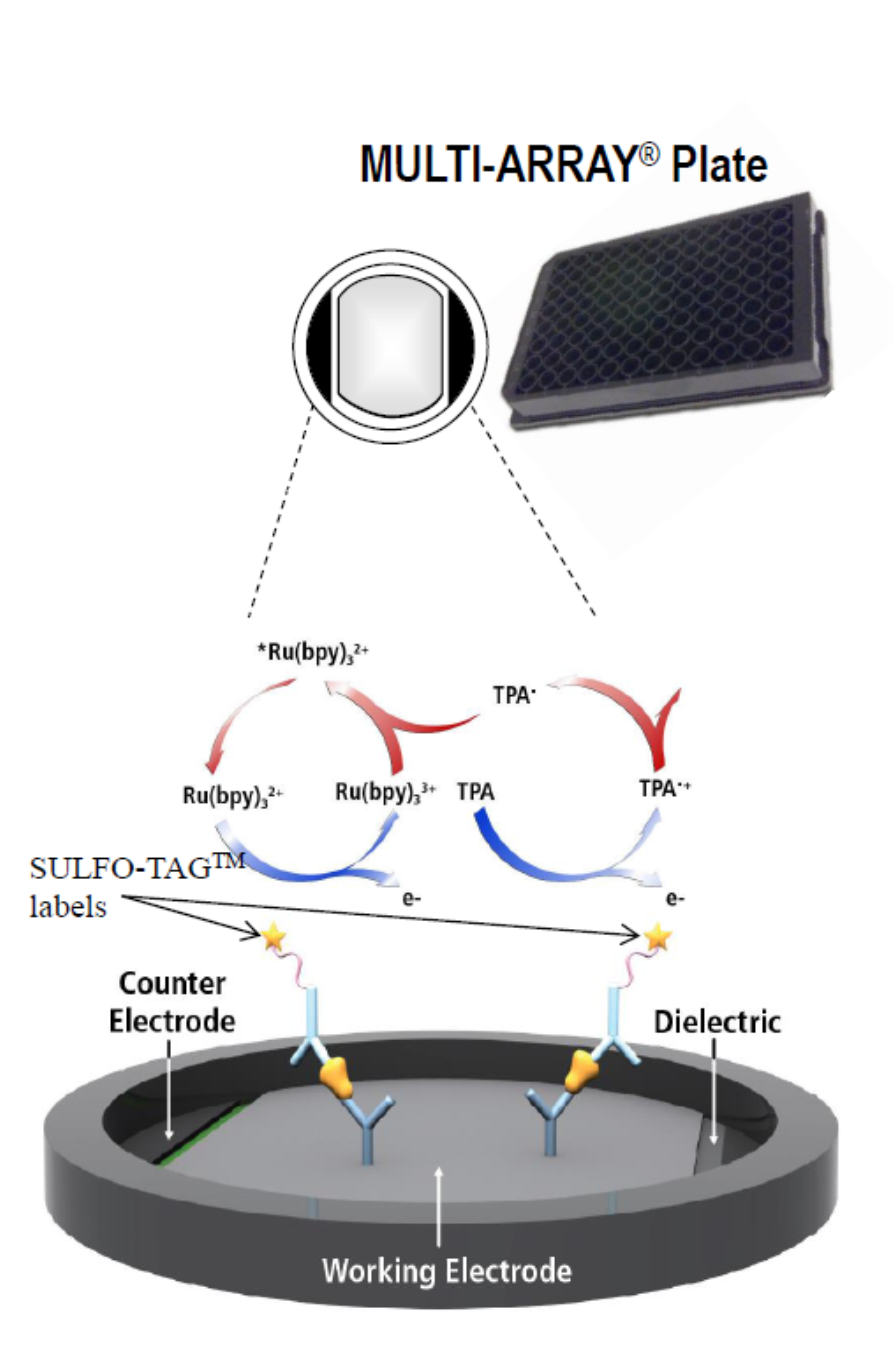
1. High binding carbon electrodes in the bottom of MULTI-ARRAY and MULTI-SPOT microplates
allow for easy attachment of biological reagents (10X greater binding capacity than polystyrene).
2. MSD assays use electrochemiluminescent labels that are conjugated to detection antibodies.
The labels are called SULFO-TAG, and allow for ultra-sensitive detection
3. Electricity is applied to the plate electrodes by an MSD instrument leading to light emission
by SULFO-TAG labels. Light intensity is then measured to quantify analytes in the sample.
Electrochemiluminescence Advantage
1. High sensitivity: Multiple excitation cycles can amplify signals to enhance light levels.
2. Broad dynamic range: The wide dynamic range of MSD-ECL detection systems means high and
low expression levels can be measured without multiple sample dilutions.
3. Low background: The stimulation method (electricity) is decoupled from the signal (light)
allowing only labels near the electrode surface to be detected.
4. Reduced assay matrix effects: Higher sample dilution factors are applied
for MSD-ECL system than conventional assays..
5. Great flexibility:
Labels are stable, non-radioactive, non-fluorescent, and conveniently
conjugated to biological molecules. In particular, fluorophore labeled cell samples
(cell lysate and culture supernatants, etc.) can be analyzed with MSD-ECL
detection system at high sensitivity.
6. Unsurpassed performance and quality: Electrochemiluminescence
is a highly successful detection system that achieves clinical quality data in a variety of sample
types, including cell supernatant, serum, plasma, and whole blood.
Ultra-Sensitive
Bead-Based Assay
Our ultra-sensitive immunoassays are established upon Quanterix’s Simoa (Single Molecule Array) technology to
detect low abundance protein targets.
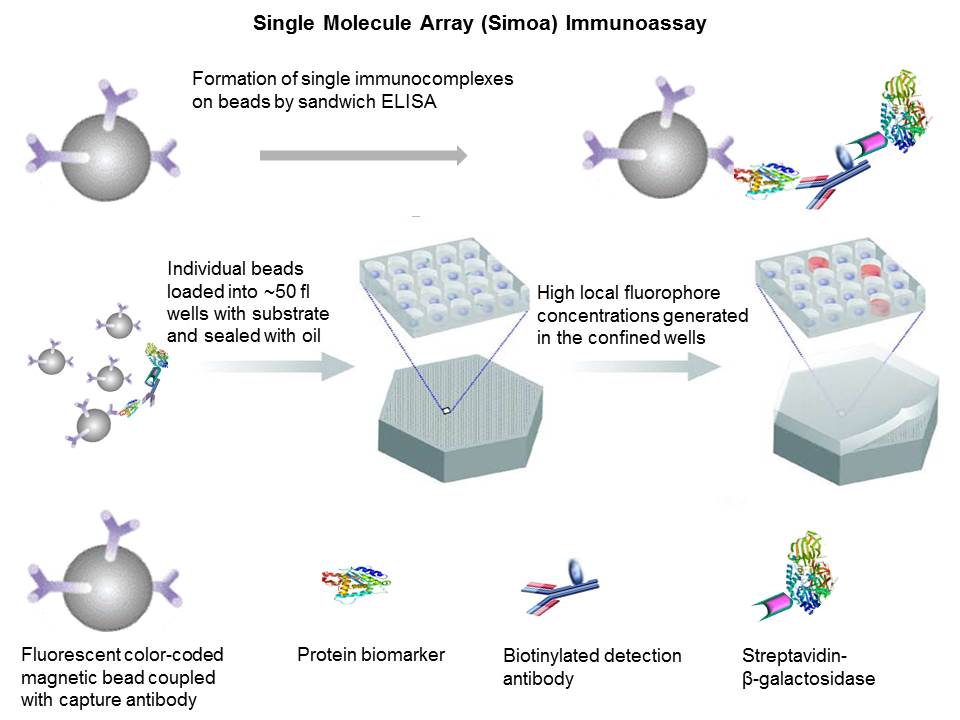
During
ultra-sensitive single molecule array (Simoa)
sandwich immunoassays, fluorescently distinct paramagnetic beads (2.7 µm
diameter) are coupled with a capture antibody. Conventional bead-based sandwich
immunoassay approach is applied but single immunocomplexes
labeled with enzyme (streptavidin-β-galactosidase) are formed on the bead surface.
When samples containing extremely low concentrations of analytes
are tested, the ratio of analyte molecules (and the
resulting immunocomplexes) to beads is small (<1) and
the percentage of beads that contain a labeled immunocomplex
follows a Poisson distribution - beads carry either a single immunocomplex or none. It is impossible to detect these low
numbers of enzyme labels using conventional detection technology, because the
fluorophores generated by each enzyme diffuse into a large assay volume
(typically greater than 0.1 mL). It takes hundreds of thousands of enzyme
labels to generate a fluorescence signal above background.
The
very low concentrations of enzyme labels on the bead surface can be detected by
loading beads (diameters of 2.7 µm) into an array of 216,000 wells (diameters
of 4.5 μm and depths of 3.25 μm)
and confining the fluorophores generated by individual enzymes to extremely
small volumes (~50 femtoliter). Beads are sealed with
oil to ensure only one bead in a well. Beads carrying a single enzyme-labeled immunocomplex generate a high local concentration of
fluorophores in the confined well. By acquiring time-lapsed fluorescence images
of the array using standard microscopic optics, it is possible to differentiate
beads associated with a single enzyme molecule (“on” well) from those not
associated with an enzyme molecule (“off” well). At low concentrations of
proteins, when the ratio of enzyme labels to beads is less than ∼1.2, beads carry either zero or low numbers of
enzymes, and protein concentration is quantified by counting the presence of
“on” or “off” beads (digital regime). At higher protein concentrations, each
bead typically carries multiple enzyme labels, and the average number of enzyme
labels present on each bead is quantified from a measure of the average fluorescence
intensity (analog regime). Both the digital and analog concentration ranges are
quantified by a common unit, namely, average number of enzyme labels per bead
(AEB). By combining digital and analog mode for fluorescence measurement of singulated beads, a linear dynamic range of over 6 orders
of magnitude to enzyme label can be achieved.
In
ultra-sensitive single molecule array (Simoa)
multiplex sandwich assays, different capture antibodies are coupled with
fluorescently distinct paramagnetic beads. Multiple analytes
in a single aliquot of serum, plasma, cell culture supernatant, cell lysate and
tissue extract are determined quantitatively and simultaneously with Quanterix’s HD-1 Simoa
Analyzer.

Since Simoa
platform enables low analyte concentrations to be
determined digitally rather than by analogue signals, it is termed a digital
immunoassay. The digital nature
of the Simoa technology allows an average of 1000×
sensitivity increase over conventional assays with CVs <10%. This
ultra-sensitivity assay platform can be completely automated with multiplexing
and custom assay capability.
Microplate-Based
Sandwich ELISA
Our conventional microplate-based immunoassay is based on a
solid phase sandwich enzyme linked immunoassay (ELISA) method. Samples,
calibrators and controls are added to the wells coated with an antibody specific
to a target. After incubation step, the target in the samples binds to the
capture antibody on the plate well. After wash step using an ELISA Plate
Washer, anti-target detection antibody conjugated with horse radish peroxidase
(HRP) is incubated in wells and binds to the target. Unbound target and HRP conjugate
are washed off by wash buffer. Upon the addition of the substrate, the
intensity of color with an ELISA plate reader is proportional to the
concentration of target in the samples. Analyte
concentrations are typically determined by 4-parametere logistic regression
algorithm with analysis of the median optical density readings of protein
standard curve.
Positive and negative controls on a plate allow for assay
quality assurance. The tests are end-point measurements.
Microplate-Based
Competitive ELISA
Another conventional microplate-based immunoassay is based
on a solid phase competitive enzyme linked immunoassays. A primary antibody is
captured by an secondary antibody coated on a
microplate. A constant concentration of biotinylated target (tracer) and varying
concentrations of unlabeled target in samples compete for binding specifically
to the primary antibody. Therefore, the concentration of tracer-bound primary antibody
is inversely proportional to the target concentrations in samples. Captured
biotinylated tracer is subsequently bound by streptavidin-conjugated
horseradish peroxidase (HRP). After washing away the unbound components, TMB is
added as a substrate for the HRP. Finally, the enzymatic reaction is terminated
by adding an acidic stop solution. The intensity of color with an ELISA plate
reader is inversely proportional to the concentration of target in the samples.
Analyte concentrations are typically determined by 4-parametere
logistic regression algorithm with analysis of the median optical density
readings of protein standard curve. Positive and negative controls on a plate
allow for assay quality assurance. The tests are end-point measurements.
Endotoxin/LPS
Measurement
Our Endotoxin/LPS test is a quantitative, kinetic assay for
the detection of Gram negative bacterial Endotoxin in a variety of samples
including serum and plasma.
A sample is mixed with the reconstituted Limulus Amebocyte Lysate (LAL) reagent, placed in the photometer or
96-well microplate and automatically monitored over time for the appearance of
turbidity. The time required before the appearance of turbidity (Reaction Time)
is inversely proportional to the amount of endotoxin present. That is, in the
presence of a large amount of endotoxin the reaction occurs rapidly; in the
presence of a smaller amount of endotoxin the reaction time is increased. The
concentration of endotoxin in unknown samples can be calculated from a standard
curve.
|
| Proenzyme |
 |
Coagulase |
|
| Coagulogen |
 |
Coagulin |
Gram negative
bacterial Endotoxin catalyzes the activation of a proenzyme in the LAL. The
initial rate of activation is determined by the concentration of Endotoxin
present. The activated enzyme (Coagulase) hydrolyzes specific bonds within a
clotting protein (Coagulogen) also present in LAL.
Once hydrolyzed, the resultant coagulin
self-associates and forms a gelatinous clot. The turbidimetric
Endotoxin/LPS assay measures the increase in turbidity (optical density) that
precedes the formation of the gel clot.
How is the LAL-Based Endotoxin/LPS testing
performed for serum/plasma?
We have built up
extensive experiences in performing Endotoxin/LPS testing for serum/plasma
samples. Preliminary sample preparation is required for serum and plasma. If
testing plasma, we use “platelet rich plasma” obtained from whole blood by
centrifuging at low speeds to remove the white and red blood cells. Serum
samples, whole blood processed to remove fibrogens,
coagulants, whole blood cells, etc., can also be tested in the same manner as
plasma.
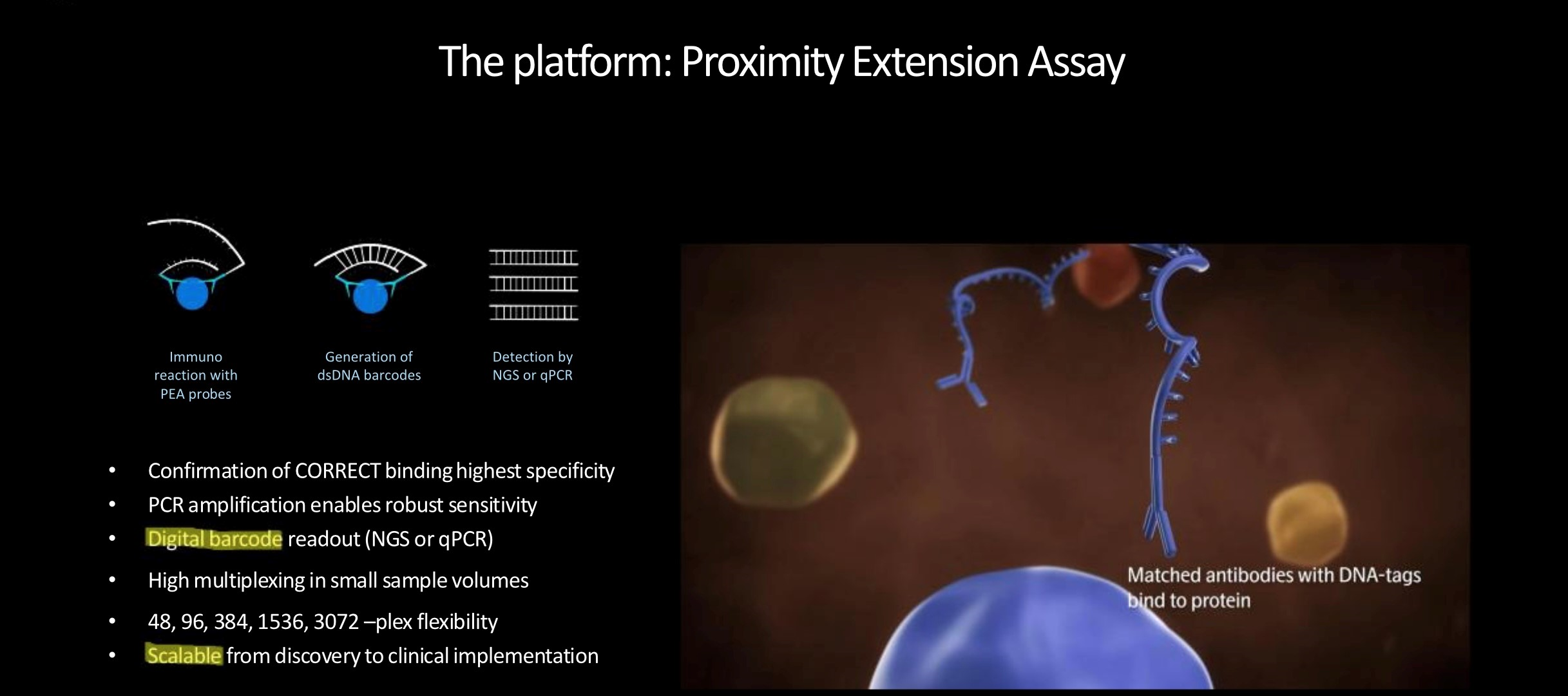
How does Olink platform work?
The unique technology behind the high-thorughput Olink multiplex assay platforms,
Proximity Extension Assay (PEA) technology, is an innovative dual recognition, DNA-coupled methodology providing exceptional readout specificity.
PEA enables high multiplex, rapid throughput biomarker analysis without compromising on data quality.
Antibody pairs labelled with DNA oligonucleotides bind target antigen in solution, allowing hybridization and extension by DNA polymerase.
This newly created piece of DNA barcode is amplified by standard PCR before transfer to an integrated microfluidic chip (IFC),
which is loaded into the instrument for qPCR and data readout.